
A rational alternative to—but not affiliated with—the “Talk.Origins Archive”
A Scientific Critique of Evolution
Dr. Lee Spetnerin an exchange with Dr. Edward E. Max
© 2000 L.M. Spetner. All Rights Reserved. [Last Modified: 7 October 2001]
r. Edward E. Max posted an essay entitled The Evolution of Improved Fitness by Random Mutation Plus Selection on http://www.talkorigins.org/faqs/fitness.html/. He asked me for my comments and, as a result, I wrote a critique of his essay (of his version updated 12 July 1999) and sent it to him on 2 August 2000. He promised me he would have it posted on the talkorigins website with a link from his essay. He responded to my critique on 22 August and I replied to his response on 29 August. I received a reply from him on 25 September that he was “looking forward to responding”, but was busy at the time. At the time of this writing (27 November 2000) I have not received any further substantive reply from him, and my comments have so far not appeared on the above website. I have therefore decided to post here a unified version of the present status of our debate. I have merged my original critique, his response, and my reply to his response to present our debate in an understandable flow. In my original critique I refer to Dr. Max in the third person. In my reply to his response, I address him in the second person.
I recommend you first read his original essay posted at the above-mentioned URL, and then read the following. I have interspersed Max’s comments into my critique where they are applicable, followed by my response to them.
At the outset, I shall establish an important and necessary guideline in this discussion of evolution. The word evolution is generally used in at least two different senses, and the distinction between them is important. On the one hand, the word evolution is used to denote the descent of all life from a putative single primitive source. It is the grand sweep of evolution that is supposed to have led from a simple beginning, something perhaps simpler than a bacterium, to all organisms living today, including humans. This descent is supposed to have occurred through purely natural means. Neo-Darwinian theory (NDT), which is the prevailing theory of evolution, teaches that this development occurred through random heritable variations in the organisms followed by natural selection. I shall denote the word evolution used in this sense as Evolution A. When evolution is discussed for popular consumption, it is most often Evolution A.
The second sense in which the word evolution is used is to denote any kind of change of a population. The change can sometimes occur in response to environmental pressure (artificial or natural selection), and sometimes it can just be random (genetic drift). I shall denote the word used in this second sense as Evolution B. Evolution B has been observed. Evolution A is an inference, but is not observable. The distinction between these two meanings of evolution parallels the distinction between macroevolution and microevolution, but the two pairs of terms are not identical. Evolution A is certainly what is called macroevolution, but what is called macroevolution is not identical with Evolution A. In any case, I prefer to use the A and B to avoid having to carry whatever baggage might go with the macro/micro distinction.
The distinction between these two meanings of evolution is often ignored by the defenders of neo-Darwinian evolution. But the distinction is critical. The claim is made for Evolution A, but the proof offered is often limited to Evolution B. The implication is that the observation of Evolution B is a substantiation of Evolution A. But this is not so. Since Evolution A is not an observable, it can only be substantiated by circumstantial evidence. This circumstantial evidence is principally the fossil record, amino-acid-sequence comparisons, and comparative anatomy. Circumstantial evidence must be accompanied by a theory of how it relates to what is to be proved. NDT is generally accepted to be that theory. The strength of the circumstantial evidence for Evolution A can therefore be no better than the strength of NDT.
The important claim of neo-Darwinism is that it can account for Evolution A. The public perceives this claim as the core of the controversy over evolution. This claim is also the source of the contention by evolutionists that life is the result of purely natural processes, which ensue from well-known natural laws. I have examined this claim in my book Not By Chance!, and have found it to be empty.
Evolution A is the principle message of evolution, namely that all life descended with modification from a putative single primitive source. The mechanism offered for the process of modification is basically the Darwinian one of a long series of steps of random variation, each followed by natural selection. The variation is generally understood today to be random mutations in the DNA. That primitive source of life is assumed to be sufficiently simple that it could have arisen from nonliving material by chance. There is no theory today that can account for such an event, but I shall refrain from addressing that issue here. That is for another place and another time. What is relevant to this discussion is that the requirement that life arose spontaneously sets, at the very least, a stringent upper limit on the complexity and information content of the putative first organism that could reproduce itself, and thus serve as a vehicle from which to launch Darwinian evolution. The issue I address here is the alleged development of all life by the neo-Darwinian process of random mutation and natural selection, starting from a sufficiently simple beginning.
Despite the insistence of evolutionists that evolution is a fact, it is really no more than an improbable story. No one has ever shown that the mechanism of NDT can result in Evolution A. Most evolutionists assume that long sequences of microevolutionary events can produce Evolution A, but no one has ever shown it to be so. (Those few evolutionists who hold that macroevolution is really different from microevolution have changed their story several times since they first came out with it, and their mechanism is so fuzzy that I have a hard time telling what it is.)
For Evolution A to work, long series of “beneficial” mutations must be possible, each building on the previous one and conferring a selective advantage on the organism. The process must be able to lead not only from one species to another, but to the entire advance of life from a simple beginning to the full complexity of present-day life. There must be a long series of possible mutations, each conferring a selective advantage on the organism so that natural selection can enable it to take over the population. Moreover, there must be not just one, but a great many such, series.
The chain must be continuous in that at each stage a change of a single base pair somewhere in the genome can lead to a more adaptive organism in some environmental context. The concept of the adaptive landscape is useful here. This concept was first introduced by Sewall Wright[1], but now nucleotide sequences of the mean population genome have taken the place of Wright’s gene combinations. There are a great many adaptive hills of various heights spread over the genomic landscape. NDT then says that it should be possible to continue to climb an “adaptive” hill to a large global maximum (or near-maximum), one base change at a time, without getting hung up on a small local maximum. No one has ever shown this to be possible.
Evolutionists often claim that if the evolutionary process were hung up on a small local adaptive maximum, a large genetic change like a recombination, or other genetic rearrangement, could bring it to another hill that has a higher peak, and place it higher up on that hill than it was before. Large adaptive changes are, however, highly improbable. They are orders of magnitude less probable than getting an adaptive change with a single nucleotide substitution, which is itself improbable. No one has shown this to be possible either.
Moreover, as I have noted in my book, the large mutations such as recombinations and transpositions are mediated by special enzymes and are executed with precision—not the sort of doings one would expect of events that were supposed to be the products of chance. Evolutionists chose the mechanism of randomness, by the way, because no one can think of any other way that beneficial mutations might occur in the absence of a law requiring them to occur. Genetic rearrangements may not be really random at all. They do not seem to qualify as the random mutations neo-Darwinists can invoke whenever needed for a population to escape from a local small adaptive maximum.
Evolutionists can argue, and rightly so, that we have no way of observing long series of mutations, since our observation time is limited to a relatively short interval. Our genetic observations over the past 100 years is thought to be more like a snapshot of evolution rather than a representative interval in which we can search for the required long series of changes. But our inability to observe such series cannot be used as a justification for the assumption that the series Darwinian theory requires indeed exist.
Max: “An equally reasonable conclusion, in my view, would be that our inability to observe such series cannot be used as a justification for the assumption that such a series of mutations did NOT occur.”Spetner: Thank you for acknowledging that what I said was reasonable. But the two statements, yours and mine, are not symmetrical. I don’t have to assume the series did not occur to make a case for the inadequacy of NDT. Those who base a theory of evolution on the occurrence of such a series are required to show that it exists, or at least that it is likely to exist. They are obliged to demonstrate an existence. I am not obliged to prove a non-existence. NDT has the convenient characteristic that the very events that would prove the theory valid are inherently not observable. Pleading that one should be excused from bringing such proofs because they are not observable does not help the evolutionist’s case. If you want to prove the theory, you had better find something observable.
Continuing my original critique, I pointed out that the argument against evolution is considerably stronger than merely noting that the evolutionists have not proved their case. It turns out that there is evidence that the series of mutations NDT requires do not, in fact, exist. The theory requires there be a vast number of possible point mutations which, coupled with natural selection, can generate the evolutionary advances that could produce Evolution A. If there really are a large number of potentially qualifying mutations, at least a few of them should have been observed in some of the many genetics laboratories around the world. All the mutations in these long series must not only confer selective advantage on the organism but they must, on the average, also contribute to the information, or complexity, increase that surely distinguishes present-day life from the putative primitive organism.
These mutations must have whatever characteristics are necessary for them to serve as elements of Evolution A. Thus, for a mutation to qualify as a representative member of the required multitude of the long series that are supposed to produce evolution, it must bring new information not just to the genome of the organism, but the information must be new to the entire biocosm.[2] The horizontal transfer of a gene from one species to another does not inject new information into the biocosm. To show evolution in action, one must at least demonstrate examples of a mutation that can serve as a prototype of those required by the theory. Such a mutation must be one that could be a contributing member of a series of mutations that could lead to the vast increase in information required by the theory. Thus, for example, a mutation that yields an enzyme new to the biocosm, or one that makes an enzyme more specific than anything in the biocosm, would be adding information. On the other hand, a mutation that disables a repressor gene causing a constitutive synthesis of an enzyme might be advantageous to an organism under special circumstances, but the disabling of a gene is not the kind of mutation the theory requires. Once in a while, such a mutation might make an adaptive contribution, but it cannot be typical of the mutations required by the theory.
Max devoted a good portion of his essay to challenging what he calls the “creationist” arguments against evolution. The arguments he challenged include false statements such as: (1) all mutations are harmful; (2) random mutations cannot increase the information content of a system; (3) the proteins had to arise by random trials without the benefit of natural selection. If he found creationists that said such things, then I suppose it’s part of the job he has assumed upon himself to refute them. His challenges, however, are hardly a telling argument for evolution. (1) Mutations have indeed been observed that confer an adaptive advantage, but that alone does not qualify them to serve as components of a series of neo-Darwinian steps. (2) Some special cases of mutations may add information to the genome, but here again, that alone does not qualify them to serve as components of an evolutionary series. (3) Although the creation of proteins by random trials is not the thesis of NDT, no one has shown that they can be generated by random mutations and natural selection in the context of evolution. His challenges are valid, but they are far from sufficient to establish NDT. I shall address these points in what follows.
The following is additional criticism of Max’s original essay that was not included in my original response.
Max challenged point (1) by indicating that beneficial mutations indeed occur. The intention of his essay was to argue for Evolution A. Had he limited himself to Evolution B, I would have no quarrel with him. He claimed that “a rare beneficial mutation can confer a survival, or reproductive, advantage to the individuals that carry it, thereby leading—over several generations—to the spread of the mutation throughout a population.”
His description is of what is often called a step in the evolutionary process. Max stated categorically that such a step can occur. Moreover, to support Evolution A, the kind of step he described must have happened over and over again, millions upon millions of times. He presented no evidence that it has ever happened, but simply tacitly assumed that it could. Can it indeed? I address that question.
One must understand that at the heart of NDT lies chance and randomness. Mutations are random events. The occurrence of a beneficial mutation at any given time in any given population is governed by chance. Even natural selection, which carries the burden of being the directive force of evolution, is subject to the laws of chance. Selection coefficients are average values. What happens in any particular instance is a random event. A mutation, even one that confers adaptive benefit on the organism, is likely to be wiped out by chance events (see Chapter 3 of my book). There is a good chance that it will disappear before it can take over the population. The question is not if it can happen, but, with what probability will it happen?
NDT is a theory that is supposed to account for the natural development of all life from a simple beginning. I don’t know why we need such a theory, because the development of life from a simple beginning is not an observable. The theory is gratuitous; it comes to account for something that was never observed.
Actually, evolutionary thinking goes like this.
- One observes present life.
- One then assumes that it arose in a natural way.
- One then concocts a theory (e.g., the NDT) to account for the observation, given the assumption.
I suppose that if the theory were really a good one, and could really explain well how life could have developed in a natural way, it would lend some credence to the assumption that life did indeed develop in a natural way. But it is not a good theory, and it does not account for what it is supposed to. Evolutionists, realizing this, have lately been reduced to arguing that if no one has a better theory that can account for the natural origin of life, then one must accept NDT. As you will see from some of Max’s comments below, he also adopts this approach. I don’t know why NDT merits the pedestal on which evolutionists have put it.
Now let’s get back to the probability of occurrence of one of those evolutionary steps of Max’s. Since they are chance events, we cannot say with any certainty that they will happen. The best we can do is to say with what probability such an event will occur. So, evolutionists have offered us a theory (NDT) that postulates a long string of random events to account for the existence of life, assuming it developed in a natural way. If the probability of those events were to turn out to be close to 1, then one could say that the theory accounts for the observation. On the other hand, if, according to the theory, the probability of those events were very low, one would have to say that the theory does not account for the observation. If a theory predicts observed events to be highly improbable, then one cannot justifiably say that the theory accounts for those events.
You would think that, since the issue of the probabilities of the evolutionary events is so crucial to the validity of the theory, the advocates of evolution would have calculated the necessary probabilities to make their case. But they haven’t. Since they have not made these calculations, Max is not entitled to assume that evolutionary steps can occur.
There is some difficulty in calculating these probabilities because the values of the relevant parameters are not all known. In my book, I addressed the problem of the probability of getting enough successful evolutionary steps to account for the evolution of the horse. In spite of the difficulties I just mentioned, I was able to calculate an important result. I found that either the probability of the horse evolving was impossibly low, or else convergent evolution cannot occur. This result refutes NDT, and with it Evolution A. Not only is Max’s point here not substantiated, it stands refuted.
Antibiotic Resistance as an Example of Evolution
Continuing his effort to show the evolutionary efficacy of beneficial mutations, Max presented in his essay the acquisition of antibiotic resistance by microorganisms as an example of evolution. He said one can “demonstrate a beneficial mutation … with laboratory organisms that multiply rapidly, and indeed such experiments have shown that rare beneficial mutations can occur. For instance, from a single bacterium one can grow a population in the presence of an antibiotic, and demonstrate that organisms surviving this culture have mutations in genes that confer antibiotic resistance.” Such an experiment shows that “de novo beneficial mutations” can arise.
My response to this is that I have shown in my book that mutations leading to antibiotic resistance fail the test of representing the mutations necessary for evolution. I summarize that argument here.
All antibiotics are derived from microorganisms. Recall the story of the serendipitous discovery of penicillin by Alexander Fleming in 1928, when he noticed that his plate of Staphylococcus bacteria was clear in the vicinity of a bread-mold contaminant. The mold was found to produce something that could lyse and kill the bacteria. That something was a molecule later named penicillin. Afterwards, other antibiotics were found to be produced by other microorganisms, such as soil bacteria. Soil has long been recognized in folk medicine as a cure for infections.
The antibiotics produced by these microorganisms serve them as a defense against attack by other microorganisms. Some microorganisms are endowed with genes that grant resistance to these antibiotics. This resistance can take the form of degrading the antibiotic molecule or of ejecting it from the cell. Unfortunately for human health care, the organisms having these genes can transfer them to other bacteria making them resistant as well. Although the resistance mechanisms are specific to a particular antibiotic, most pathogenic bacteria have, to our misfortune, succeeded in accumulating several sets of genes granting them resistance to a variety of antibiotics.
The acquisition of antibiotic resistance in this manner qualifies as evolution only in the sense that it is an adaptive hereditary change. It is an example only of Evolution B. It is not the type of evolution that can make a baboon out of a bacterium. The genetic change is not the kind that can serve as a prototype for the mutations needed to account for Evolution A. The genetic changes that could illustrate the theory must not only add information to the bacterium’s genome, they must add new information to the biocosm. The horizontal transfer of genes only spreads around genes that are already in some species.
It turns out, however, that a microorganism can sometimes acquire resistance to an antibiotic through a random substitution of a single nucleotide, and this is the kind of example Max presented. Streptomycin, which was discovered by Selman Waksman and Albert Schatz and first reported in 1944, is an antibiotic against which bacteria can acquire resistance in this way. But although the mutation they undergo in the process is beneficial to the microorganism in the presence of streptomycin, it cannot serve as a prototype for the kind of mutations needed by NDT. The type of mutation that grants resistance to streptomycin is manifest in the ribosome and degrades its molecular match with the antibiotic molecule. This change in the surface of the microorganism’s ribosome prevents the streptomycin molecule from attaching and carrying out its antibiotic function. It turns out that this degradation is a loss of specificity and therefore a loss of information. The main point is that Evolution A cannot be achieved by mutations of this sort, no matter how many of them there are. Evolution cannot be built by accumulating mutations that only degrade specificity.
In the final paragraph of my original critique, I said the following:
The mutations needed for macroevolution have never been observed. No random mutations that could represent the mutations required by NDT that have been examined on the molecular level have added any information. The question I address is: Are the mutations that have been observed the kind the theory needs for support? The answer turns out to be NO! Many have lost information. To support NDT one would have to show many examples of random mutations that add information. Unless the aggregate results of the genetic experiments performed until now is a grossly biased sample, we can safely dismiss neo-Darwinian theory as an explanation of how life developed from a single simple source.
Max: “I think that the sample of genetic mutations you cite is in fact biased, incorrectly interpreted, and much too small and non-systematic to draw such a sweeping conclusion. I will try to explain why I believe this.“Some streptomycin resistance mutations do, as you point out, reflect mutations of the ribosomal protein S12 which cause loss of binding of this antibiotic, which you interpret as ‘loss of information.’ However, you ignore other mutations of this protein that do not lead to loss of antibiotic binding (e.g. Timms et al., Mol Gen Genet 232:89, 1992). According to your formulation, these mutations would not represent a loss of information, yet they are represent natural mutations that are adaptive under conditions of exposure to streptomycin. Would you accept that this kind of mutation is a good model for an adaptive evolutionary change consistent with neo-Darwinian Theory?
“Using your own example of streptomycin resistance, I have pointed out that some mutations of the S12 ribosomal protein do not represent a ‘loss of information’ even by your own questionable criteria.”
Spetner: First of all, I would recommend that you not refer to my criteria of information loss as “questionable” until you understand them. See below, where I explain your misunderstanding. You let your own tacit assumptions get in the way of understanding my thesis.
Furthermore, you misunderstood the paper by Timms et al., which you cited. All of the adaptive mutations reported in that paper show reduced binding of the streptomycin molecule. The 12 adaptive mutations reported in the S12 protein fall into two categories. There was no example of what you claimed I ignored. Five of those mutants are designated as streptomycin resistant (Smr), and seven are designated as streptomycin dependent (Smd). All 12 of them, in the words of the authors “reduce the affinity of the ribosome for streptomycin.” Perhaps you would like to point out to me where in that paper they mention mutations in S12 do not lead to reduced binding, and which you claim I have ignored.
Max: “… how about the single amino acid substitution in a blowfly carboxylesterase that converts this enzyme into an organophosphorus hydrolase under selection by organophosphate insecticides [Newcomb et al., PNAS 94:464, 1997]?”Spetner: In the Newcomb et al. paper that you cited, the experimenters started with 15 existing strains of blowflies, some of which were resistant to organophosphorus (OP) insecticide and some were not. No mutations were imposed or observed. The amino-acid differences between the two groups were only assumed to have arisen by mutation.
The esterase enzyme E3 plays an important role in the operation of the fly’s nervous system. OP insecticides kill the insects by interfering with this activity. Newcomb et al. found that the resistant allele of the gene encoding the enzyme E3 and the corresponding susceptible allele differ from each other in 19 nucleotides. These differences translate into 5 amino-acid differences between the enzymes. The authors concluded from their study that one of those 5, namely Gly137® Asp, could account for both a loss of esterase activity and an acquisition of OP hydrolase activity.
Although it is not certain that the difference in the activities of this enzyme arose through a random mutation, let us even suppose it did. If it did, then this mutation is not likely to have occurred recently, because much time would be needed to have accumulated all the 19 nucleotide differences between these two phenotypes. Both phenotypes have likely been in the populations of blowflies long before OP insecticides entered the environment. The resistant allele must therefore have adaptive advantages in addition to OP insecticide resistance.
One can say with a large measure of confidence that the resistant strain did not arise through a single random mutation and proliferate through natural selection in the presence of OP. This is evident from a close examination of Fig. 2 of the above-cited paper. From what the authors have shown, if there were such a mutation it would have been the substitution Gly137® Asp. The authors created two chimeric alleles of the E3 enzyme. One of these (let’s call it the susceptible chimera) had Gly137, corresponding to the allele susceptible to diazinon insecticide, but had the other 4 of the 5 discordant amino acids identical with the wild-type allele resistant to diazinon. The other chimera (we’ll call it the resistant chimera) had the opposite; it had Asp137, and the other 4 of the 5 discordant amino acids identical with the susceptible allele. The authors measured the OP hydrolase activity in the wild-type susceptible allele, the susceptible chimera, the wild-type resistant allele, and the resistant chimera. They presented the results of their measurements in Fig. 2 of their paper, which shows the following:
- There is negligible OP hydrolase activity in the wild-type susceptible allele of E3 and in the susceptible chimera.
- There is marked OP hydrolase activity in the wild-type resistant allele of E3 and in the resistant chimera. It is this activity that the authors understand to be responsible for the resistance to OP insecticide.
- The OP hydrolase activity of the resistant chimera is about two and a half times that of the wild-type resistant allele.
Accepting the authors’ premise that the OP hydrolase activity is responsible for the resistance, we can say that a strain of blow fly whose E3 enzyme is identical with that of the wild-type susceptible, except for the single substitution Asp137, should be even more resistant than the wild-type resistant strain. Therefore, if a mutation occurred in the susceptible strain achieving the substitution Gly137® Asp, it should be more adaptive than the wild-type susceptible strain in the presence of diazinon insecticide. That being the case, the resistant population of blow flies should have an E3 enzyme with only Asp137 differing from that of the wild-type susceptible strain. Since that is not the case, one can conclude that the other 4 of the discordant amino acids must have some overriding adaptive value that trumps the greater OP hydrolase activity of the resistant chimera, even though we don’t know what that adaptive value is. In light of these data, one can conclude that the substitution Gly137® Asp did not arise by random mutation from the susceptible strain.
Moreover, to tell if the substitution, Gly137® Asp, even if it did arise by a random mutation, represents an addition or a loss of information, we must know more about how the mutation affects the enzyme’s hydrolase activity on more than just the one substrate. As in the example I showed in my book (and described briefly below), what looked like an enhancement of activity on one substrate, coupled with a degradation of activity on another, turned out to be nothing more than a simple reduction of specificity of the enzyme over wider a set of substrates. As I wrote in my original comments (see below) on your posting, one must be careful about jumping to conclusions about what constitutes an information increase. This is not a weasel statement. One can know when one has enough data to make a judgment. If the activity profile of the mutant enzyme over several different substrates sharpens by increasing the activity on one substrate and concomitantly decreasing the activity on other substrates, there is an increase in selectivity and hence an increase in information. If, on the other hand, the activity profile of the mutant enzyme over a set of substrates is flatter than that that of the wild type, then information has been lost. One just needs enough data to be able to see an activity profile over several substrates for both the mutant and the wild type.
Max: “Certainly you are not correct when you say ‘all known examples of these mutations lose information rather than gain it.’”Spetner: Since you have not shown any valid counterexamples, my statement still stands, and your statement falls. None of the examples you gave qualifies as a random mutation in the germ line that could be typical of those required for Evolution A. The context in which I made the above statement was that of random mutations in the germ line that, according to NDT, are capable of producing Evolution A.
You must admit that the most widely used examples by evolutionists to show evolution in action do in fact lose information. You have used such an example yourself in your posted essay—the evolution of antibiotic resistance in microorganisms. These so-called “best examples” are poor and do not demonstrate, nor even indicate a typical contribution to, Evolution A.
You failed in your attempt to rebut my statement that all known examples of random mutations that could play a role in Evolution A lose information rather than gain it.
Evolution and the Increase of Information
In my critique, I included for pedagogical purposes the following short explanation of the information in enzymatic activity and its measurement:
I shall emphasize again: There is no theorem requiring mutations to lose information. I can easily imagine mutations that gain information. The simplest example is what is known as a back mutation. A back mutation undoes the effect of a previous mutation. If the change of a single base pair in the genome were to change to another and lose information, then a subsequent mutation back to the previous condition would regain the lost information. Since these mutations are known to occur, they form a counterexample to any conjecture that random mutations must lose information. An important point I make in my book, and which I emphasize here, is that, as far as I know, no mutations observed so far qualify as examples of the kind of mutations required for Evolution A.
In discussing mutations in my book I noted in each case in which the molecular change was known, that it could not serve as a prototype for the mutations required by NDT. In all the cases I discussed, it was the loss of information that prevented the mutation from serving as a prototype of those required by NDT. The back mutation likewise cannot serve as a prototype of the NDT-required mutations. Here, the reason is not that it loses information—it actually gains information. But the information it gains is already in the biocosm and the mutation contributes nothing new. Evolution is not accounted for if the only information gain was by back mutations.
In my book, I did not quantify the information gain or loss in a mutation. I left it out mainly because I was reluctant to introduce equations and scare off the average reader. And anyway, I thought it rather obvious that a mutation that destroys the functionality of a gene (such as a repressor gene) is a loss of information. I also thought it rather obvious that a mutation that reduces the specificity of an enzyme is also a loss of information. But I shall take this opportunity to quantify the information difference before and after mutation in an important special case, which I described in my book.
The information content of the genome is difficult to evaluate with any precision. Fortunately, for my purposes, I need only consider the change in the information in an enzyme caused by a mutation. The information content of an enzyme is the sum of many parts, among which are:
- Level of catalytic activity
- Specificity with respect to the substrate
- Strength of binding to cell structure
- Specificity of binding to cell structure
- Specificity of the amino-acid sequence devoted to specifying the enzyme for degradation
These are all difficult to evaluate, but the easiest to get a handle on is the information in the substrate specificity.
To estimate the information in an enzyme I shall assume that the information content of the enzyme itself is at least the maximum information gained in transforming the substrate distribution into the product distribution. (I think this assumption is reasonable, but to be rigorous it should really be proved.)
We can think of the substrate specificity of the enzyme as a kind of filter. The entropy of the ensemble of substances separated after filtration is less than the entropy of the original ensemble of the mixture. We can therefore say that the filtration process results in an information gain equal to the decrease in entropy. Let’s imagine a uniform distribution of substrates presented to many copies of an enzyme. I choose a uniform distribution of substrates because that will permit the enzyme to express its maximum information gain. The substrates considered here are restricted to a set of similar molecules on which the enzyme has the same metabolic effect. This restriction not only simplifies our exercise but it applies to the case I discussed in my book.
The products of a substrate on which the enzyme has a higher activity will be more numerous than those of a substrate on which the enzyme has a lower activity. Because of the filtering, the distribution of concentrations of products will have a lower entropy than that of substrates. Note that we are neglecting whatever entropy change stems from the chemical changes of the substrates into products, and we are focusing on the entropy change reflected in the distributions of the products of the substrates acted upon by the enzyme.
The entropy of an ensemble of n elements with fractional concentrations f1,…,fn is given by
and if the base of the logarithm is 2, the units of entropy are bits.
(1) As a first illustration of this formula let us take the extreme case where there are n possible substrates, and the enzyme has a nonzero activity on only one of them. This is perfect filtering. The input entropy for a uniform distribution of n elements is, from (1), given by
since the fi's are each 1/n. The entropy of the output is zero,
(2) because all the concentrations except one are zero, and the concentration of that one is 1. Then the decrease in entropy brought about by the selectivity of the enzyme is then the difference between (2) and (3), or
(3) Another example is the other extreme case in which the enzyme does not discriminate at all among the n substrates. In this case the input and output entropies are the same, namely
Therefore, the information gain, which is the difference between HO and HI, in this case is zero,
(4) We normalize the activities of the enzyme on the various substrates and these normalized activities will then be the fractional concentrations of the products. This normalization will eliminate from our consideration the effect of the absolute activity level on the information content, leaving us with only the effect of the selectivity.
(5) Although these simplifications prevent us from calculating the total entropy decrease achieved by action of the enzyme, we are able to calculate the entropy change due to enzyme specificity alone.
The Dangers of Conclusion Jumping
As a final example let me take part of a series of experiments I discussed in my book, which demonstrate the dangers of conclusion jumping. This subject bears emphasis because evolutionists from Darwin on have been guilty of jumping to unwarranted conclusions from inadequate data. I shall here take only a portion of the discussion in my book, namely, what I took from a paper by Burleigh et al.[3] to illustrate my point.
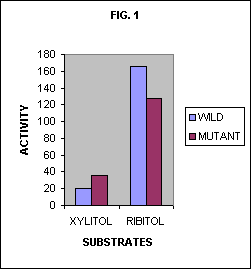
Ribitol is a naturally occurring sugar that some soil bacteria can normally metabolize, and ribitol dehydrogenase is the enzyme that catalyzes the first step in its metabolism. Xylitol is a sugar very similar in structure to ribitol, but does not occur in nature. Bacteria cannot normally live on xylitol, but when a large population of them were cultured on only xylitol, mutants appeared that were able to metabolize it. The wild-type enzyme was found to have a small activity on xylitol, but not large enough for the bacteria to live on xylitol alone. The mutant enzyme had an activity large enough to permit the bacterium to live on xylitol alone.
Fig. 1 shows the activity of the wild-type enzyme and the mutant enzyme on both ribitol and xylitol. Note that the mutant enzyme has a lower activity on ribitol and a higher activity on xylitol than does the wild-type enzyme. An evolutionist would be tempted to see here the beginning of a trend. He might be inclined to jump to the conclusion that with a series of many mutations of this kind, one after another, evolution could produce an enzyme that would have a high activity on xylitol and a low, or zero, activity on ribitol. Now wouldn’t that be a useful thing for a bacterium that had only xylitol available and no ribitol? Such a series would produce the kind of evolutionary change NDT calls for. It would be an example of the kind of series that would support NDT. The series would have to consist of mutations that would, step by step, lower the activity of the enzyme on the first substrate while increasing it on the second.
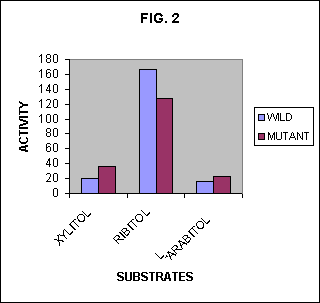
But Fig. 1 is misleading in this regard because it provides only a restricted view of the story. Burleigh and his colleagues also measured the activities of the two enzymes on another similar sugar, L-arabitol, and the results of these measurements are shown in Fig. 2. With the additional data on L-arabitol, a different picture emerges. No longer do we see the mutation just swinging the activity away from ribitol and toward xylitol. We see instead a general lowering of the selectivity of the enzyme over the set of substrates. The activity profiles in Fig. 2 show that the wild-type enzyme is more selective than is the mutant enzyme.
In Fig. 1 alone, there appears to be a trend evolving an enzyme with a high activity on xylitol and a low activity on ribitol. But Fig. 2 shows that such an extrapolation is unwarranted. It shows instead a much different trend. An extrapolation of the trend that appears in Fig. 2 would indicate that a series of such mutations could result in an enzyme that had no selectivity at all, but exhibited the same low activity on a wide set of substrates.
The point to be made from this example is that conclusion jumping from the observation of an apparent trend is a risky business. From a little data, the mutation appears to add information to the enzyme. From a little more data, the mutation appears to be degrading the enzyme’s specificity and losing information.
Just as we calculated information in the two special cases above, we can calculate the information in the enzyme acting on a uniform mixture of the three substrates for both the wild type and the mutant enzyme. Using the measured activity values reported by Burleigh et al. we find the information in the specificities of the two enzymes to be 0.74 and 0.38 bits respectively. The information in the wild-type enzyme then turns out to be about twice that of the mutant.
The evolutionist community, from Darwin to today, has based its major claims on unwarranted conclusion jumping. Darwin saw that pigeon breeders could achieve a wide variety of forms in their pigeons by selection, and he assumed that the reach of selection was unlimited. Evolutionists, who have seen crops and farm animals bred to have many commercially desirable features, have jumped to the conclusion that natural selection, in the course of millions of years, could achieve many-fold greater adaptive changes than artificial selection has achieved in only tens of years. I have shown in my book that such extrapolations are ill founded because breeding experiments, such as those giving wheat greater protein content or vegetables greater size, result from mutations that disable repressor genes. The conclusions jumped to were false because they were based on data that could not be extrapolated to long sequences. One cannot gain information from a long sequence of steps that all lose information. As I noted in my book, that would be like the merchant who lost a little money on each sale, but thought he could make it up on volume.
Max: “I want to make it clear that I don’t buy your interpretation of certain specific mutations as reflecting a ‘loss of information.’ You state that the ‘information content of an enzyme is the sum of many parts, among which are: level of catalytic activity, specificity with respect to the substrate, strength [and specificity] of binding to cell structure, [and] specificity of the amino-acid sequence devoted to specifying the enzyme for degradation.’ This formulation is vague, non-quantitative, not supported by clear logic, not accepted in the scientific literature (to the best of my knowledge; please educate me if I am wrong), and in my view not useful.”Spetner: Ed, the level of your argument here is quite low. You have seen this entire section (above), and you took from the introduction my list of what characteristics can contribute to the information content of an enzyme and criticized it for being non-quantitative (followed by other pejorative epithets). Is that supposed to be some sort of debating tactic? In any case, the tactic is out of place in this discussion. From the context of what I wrote, it should have been clear to you that this partial list of characteristics that can contribute to the information in an enzyme was an introduction to my quantitative estimate of one of the characteristics of specificity of an enzyme. After I showed how one might calculate the information related to a type of specificity, I showed how a mutation that appeared to enhance activity on a new substrate actually reduced the information by about 50%.
It is elementary that specificity translates into information and vice versa. Have you ever played 20 questions? With the YES/NO answers to 20 judicious questions, one can discover a previously-chosen number between 1 and a million. If the questions are well chosen, those YES/NO answers can be worth one bit of information each, and 20 bits can specify one object out of a million. Twenty bits of information translates to specificity of one part in a million. Ten bits—to one part in a thousand.
The Zip codes in the US also demonstrate that specificity and information are two sides of the same coin and go hand in hand. An address in the United States can be completely specified by the nine-digit zip code. One digit of information will narrow down the address from being anywhere in the United States to being in just a few states. Thus if the first digit is a 6, the address is located somewhere in Illinois, Missouri, Kansas, or Nebraska.
A second digit of information will add specificity by narrowing down the address further. A 3, 4, or 5 in the second digit puts the address in Missouri. A 3 in the second digit puts it in the eastern portion of the state. Two digits of information are more specific than one.
A third digit of information is still more specific, narrowing down the address even more, making it still more specific. If the third digit is a 1, the address is specific to St. Louis and its suburbs. The next two digits of information pin down the address to within a few blocks. The last 4 digits of information can locate a specific building. Thus, it is clear that the information contained in the digits of the zip code translate into specificity.
There is no question about it: SPECIFICITY = INFORMATION.
Max: “… there are several other ways of considering how mutations affect information. In my view, even if all S12 mutations that caused streptomycin resistance abolished antibiotic binding, a reasonable argument could still be made that such mutations represent a gain of information rather than a loss. In the universe of all the possible S12 amino acid sequences that can function in the ribosome, essentially all S12 proteins found in ‘wild-type’ bacteria (i.e., those grown in the absence of streptomycin) bind to this antibiotic. The S12 sequences that allow bacterial growth in the presence of streptomycin represent a small subset of the universe of observed functional S12 sequences. Therefore by growing bacteria in streptomycin we select for a specific and small subset of possible S12 sequences; thus it might be argued that we have forced a small increase the information content of the genome by narrowing the choice of S12 sequences.”Spetner: I cannot agree with what you wrote here. The wild-type S12 proteins that bind to the streptomycin molecule also form a subset of the universe of all possible S12 proteins. The set of S12 proteins that allow bacterial growth in streptomycin (i.e. that do not bind to the antibiotic) form a disparate subset of the universe of S12 proteins. My intuition tells me that the set that binds (the susceptible set) is smaller, and therefore has a smaller entropy, than the set that does not bind (the resistant set). Mutations that appear in the presence of the antibiotic convert one subset to the other. A mutation that transfers the enzyme from a low-entropy set to a higher-entropy set loses information; it does not gain it.
Max: “Alternatively, it could just as well be argued that in all cases of single amino acid replacements there has been no change in information content at all, in that any given amino acid sequence is equally ‘improbable’ compared with any other amino acid sequence of the same length.”Spetner: This is not a useful concept. It is like the pleading of the poker player who had a bust hand. When it came to the call, his opponent showed four aces. He pleaded that his bust hand was just as improbable, and therefore worth as much, as the four-aces, and suggested they split the pot. He’s right about the probabilities of the two hands, but in the context of poker, four aces win and the bust hand loses. Although in the context of the organism’s survival in streptomycin, the degraded specificity of the S12 protein is beneficial, in the context of evolution, it is a dead end and it loses.
Max: “Certainly you have provided no theoretical justification for using your arbitrary criteria such as ‘specificity of binding’ to assess information content; indeed, you fail to provide any quantitative theory of how all the criteria you list (‘level of catalytic activity, specificity with respect to substrate, . .’ etc) would be integrated into a quantitative information measure.”Spetner: On the contrary, I have provided substantial theoretical justification for equating information to specificity. You just chose to ignore what I wrote.
Max: “In general, if a protein has evolved under selection for a specific function, changes in the structure of that protein to meet some new criterion can be expected to adversely affect the original function. This is true in ribosomal S12 proteins that have become streptomycin resistant (they are less efficient in proof-reading) and is clear in the example of the carboxylesterase, which loses this activity essentially completely when mutated to become an organophosphorus hydrolase. The structure of any protein—like the product of engineering design—involves trade-offs between various opposing optimization ‘goals’. Thus it is likely that intense selection for resistance to a lethal agent—exactly the kind of quick experimental protocol useful for laboratory models of adaptive evolutionary change—will lead to mutations that involve what might be construed by you as a ‘loss of information’; something is always likely to be lost when a modified, mutated protein becomes prevalent in the face of a new selective pressure. This fact explains, I believe, why such genetic experiments may in fact be ‘grossly biased’ in the way that led you to inappropriately dismiss neo-Darwinian theory.”Spetner: You show here that you misunderstand what I mean by a mutation losing information. If a mutation in an enzyme were to lose its specificity to one substrate and gain specificity to another substrate, I would credit the mutation with a gain of information and I would not ‘construe’ it as a loss. But I will not credit it with a gain if the enzyme increased its activity to another substrate merely by becoming less specific, as in the example I gave above with ribitol and xylitol.
What you have presented is not so much a case for bias as it is a pleading that any modification to a protein must cause some degradation, and therefore you want to be excused from having to show a case where information is increased. But you have overplayed your hand. You seem to be saying in effect that because proteins have evolved so well, any change will degrade them. (If that were so, it would be a good argument for Creation.)
Suppose a mutation causes a protein to become more adaptive in a particular environment. Then by your thesis, it is already so well evolved that “something is always likely to be lost when a modified, mutated protein becomes prevalent in the face of a new selective pressure.” You imply that the loss is one of information, because that’s the context of this discussion. But then, according to you, after that modification, it is again well evolved, so the next time it undergoes an adaptive mutation, it must again lose something. Continuing the process you have described, the protein will continue to lose something. You have just consigned the evolutionary process to a dead end!
Max: “But consider this: If blowflies happened to have duplicated their carboxyhydrolase gene before they were exposed to organophosphates, and if they mutated one of their two copies to organophosphate hydrolase, we would have a clear example of an increase in genetic information: creation of a new functional gene … without any loss of information since the original sequence would be intact in the unaltered copy.”Spetner: I have already shown above that the organophosphorus hydrolase activity did not necessarily come from a single point mutation. I have also noted that we don’t have enough information to know if the acquisition of this activity is a loss or a gain of information. Furthermore, you don’t have to keep bringing up the necessity of gene duplication. If an enzyme lost its old activity to gain a new specificity, I would credit it with a gain of information without regard to the loss of the old activity. I have always assumed that gene duplication is available to evolution.
Max: “Now, gene duplications are rather rare events, and favorable mutations are also rare; so the combined frequencies of these two events are so rare that they are not likely to be observable in a laboratory experiment. But if we look at many gene systems in modern animals we can see how they might have been caused by duplication followed by mutation to a new, or at least slightly different, function.”Spetner: As I said above, I grant the possibility of gene duplication, so you needn’t throw that in to make the probability low. If I saw the gain of specificity through a random mutation, I would credit the mutation with an increase of information without deducting for the loss of the old activity. A single point mutation (which is all that NDT requires at each step) is not very rare considering all the genetic experiments that have been performed throughout the world. If there really are as many adaptive, information-adding mutations as NDT needs, we should expect to have seen many of them.
Max: “As an example of such a system, let’s consider a gene locus that I have studied in my lab: the human immunoglobulin heavy chain (or IgH) locus. In the human locus one sees evidence of a large DNA duplication that created two copies that are highly similar in both coding and non-coding flanking regions. One duplicate includes constant region sequences known as gamma3, gamma1, pseudo-epsilon and alpha1, while the second copy contains gamma2, gamma4, epsilon and alpha2. More primitive primates like the New World monkeys appear to have a single copy of this locus and a single gamma gene. The four human gamma chain genes are thus thought to have derived from a single ancestral gamma chain gene in a primate ancestor by a series of duplications and mutations.… In the ancestral primate we had one non-specialized gene whereas in modern humans we have four specialized genes. This is exactly the sort of genetic change that would be consistent with neo-Darwinian evolution leading to an increase in complexity.”Spetner: Yes, information would have been increased if what you speculate had indeed happened. The proof would only lie in showing that it has indeed happened through random mutations and natural selection. Let us not lose sight of the requirement of neo-Darwinian evolution for long series of single-nucleotide substitutions, where each mutation makes the phenotype sufficiently more adaptive than it was to permit the mutated phenotype to take over the population through natural selection with a high probability. It is far from clear that the individual mutations you suggest will each be adaptive and selected at each step. You cannot show this—you merely assume it. You are postulating an historical event that cannot possibly be verified. It seems that all of your arguments are based on postulating events that are inherently not observable. That should make one a little suspicious of the theory, shouldn’t it?
Max: “I realize that the above model for the human IgH locus is hypothetical and assumes that the evolutionary triad of duplication, random mutation and selection is a reasonable naturalistic explanation for the four human gamma genes. We cannot verify this explanation since we can never know the properties of the primordial ancestral gamma immunoglobulin, or know the series of mutations that occurred in the various duplicate gamma genes during our evolution from that primordial ancestor. What I am asking is: is there anything so implausible in this model to justify your suggestion that we should ‘dismiss neo-Darwinian theory’ as an explanation for this example?”Spetner: Yes, it is implausible because you are postulating a series of events of a type for which there is evidence that they have not occurred. If they had occurred to produce Evolution A, there should have been a vast number of them, and there aren’t. Had there been the required large number of them, we should have seen some of them in all the genetic experiments performed in all the laboratories of the world. And we haven’t, to my knowledge, seen a single one.
Max: “Or more to the point, exactly what alternative explanation for the origin of the four human gamma genes do you propose that is more plausible than the one I offered?”Spetner: How does Creation grab you? You probably are reluctant to admit that possibility, but you can think of it as a default position. It cannot be demonstrated scientifically, not because of any philosophical defect in the proposition, but because of the limitations of Science. Because Science is incapable of dealing with it does not mean it hasn’t happened. There are, after all, some truths in the physical world that cannot be reached by Science, just as there are mathematical truths that cannot be reached by mathematical proof.[4] If we don’t have a scientifically viable theory to account for the origin of the four human gamma genes, or for the origin of life itself, we needn’t despair. Not every mystery necessarily has a scientific solution. I do not mean to say that one should not look for a scientific solution. One should. But not having such a solution is not a license to make up stories and pass them off to a gullible public as Science. Because I don’t have a (scientifically) ‘plausible’ explanation of the origin of life, does not mean that your improbable stories are correct and should be foisted on the public under the guise of scientific truth.
Max: “This is important, because considering the weaknesses I have pointed out in your arguments, you are far from having definitively ruled out the neo-Darwinian evolutionary triad as the correct explanation for what you call the ‘grand sweep of evolution’”[I am now calling this Evolution A (LMS)].Spetner: As you can see from my above remarks, you have not succeeded in pointing out any weakness in my arguments. What you call the “Darwinian evolutionary triad” is no more than a big bluff. It has great theoretical and empirical difficulties, which neither you nor anyone else has succeeded in overcoming.
Mutations in the Immune System
Max’s field of expertise is the immune system. This is the field in which he does research and in which he has published. In his original posting, his pièce de résistance was the presentation of somatic mutations in B lymphocytes (B cells) of the vertebrate immune system as examples of random mutations that add information. He implied that Evolution A could follow this method to achieve baboons from bacteria. I agree with him that these mutations add information to the B-cell genome. I also agree that they are random. They are random, however, only in the base changes they make; they are not random in where in the genome they can occur. More important, I do not agree that the Evolution A could be achieved through such mutations, and I shall show why.
Although the somatic mutations to which Max referred are point mutations that do indeed add information to the genome of the B cells, they cannot be applied to Darwinian evolution. These are not the kind of mutations that can operate as the random mutations required by NDT, which allegedly can, through chance errors, build information one base change at a time.
For one thing, the rate of the somatic mutations in the immune system is extremely high—more than a million times normal germ-line mutation rates. For this reason they are called hypermutations. If an organism had a germline mutation rate that was even a small fraction of this rate it could not survive. For a second thing, the hypermutations in the B cells are restricted to a specific tiny portion of the genome, where they can do no harm but only good. The entire genome of the B cell could not mutate at this rate; the hypermutation must be restricted only to the region encoding selected portions of the variable part of the antibody.
The mutation rate of the hypermutating part of the B cell’s genome is usually about 10-3 per base pair per replication[5], and it can be as high as 10-2 per base pair per replication[6]. These rates cannot produce Darwinian evolution. If a genome were to mutate at this rate, there would be, on the average, several mutations in every gene, with a high probability that many of them would be fatal for the organism. Darwinian evolution could not occur with such rates.
These high rates are essential for the working of the immune system. In each replication of a B cell, about 30 of the 300 or so gene regions encoding the CDRs (complementarity-determining regions) will have a mutation. A lower mutation rate would make for a less efficient immune system. The high mutation rates, so necessary for the immune system, if applied to an entire organism for evolutionary purposes, would be fatal many times over.
Note that these hypermutations are limited to a restricted portion of the genome. Moreover, the hypermutations are mediated by special enzymes. Although the hypermutations are random in the changes they make in the bases of the genome, they are not random in the positions in which they occur. They occur only in the small region in which they are needed, and occur there through enzymes that apparently play only that role. Furthermore, they occur only when they are switched on by the controlling mechanism of B-cell maturation. Thus, it is clear that the hypermutations in B cells cannot serve as a prototype for the random mutations required for NDT.
Max: “You … declare that the B cell example is a poor model for what happens in ‘Darwinian’ evolution, and you cite two reasons: (1) the mutation rate in this model is much higher than what is seen in non-immunoglobulin genes and in non-B-cells; and (2) these ‘hypermutations’ are mediated by ‘special enzymes.’ With regard to your first point, I agree that the mutation rate is higher in the B cell example than in evolution, but I fail to see why that fact weakens the usefulness of the example as a model for evolution. If adaptive mutations that increase information in the genome of a B lymphocyte population can occur over one week given a high mutation rate, what theoretical argument would lead you to reject the idea that adaptive mutations that increase information in the genome of a germ cell population could occur over many millions of years given a much lower mutation rate?”Spetner: The theoretical argument hinges on the fact that the benefit that accrues to the immune system is a nonlinear function of the mutation rate. Evolution requires a long series of steps each consisting of an adaptive mutation followed by natural selection. In this series, each mutation must have a higher selective value than the previous[7]. Thus, the evolving population moves across the adaptive landscape always rising toward higher adaptivity. It is generally accepted that the adaptive landscape is not just one big smooth hill with a single maximum, but it is many hills of many different heights. Most likely, the population is on a hill that is one of the many lowest and not on one of the few highest in the landscape. It will then get stuck on a low local maximum of adaptivity and will not be able to move from it. That is particularly likely because the steps it takes are very small—only one nucleotide change at a time. The problem is compounded by the lack of freedom of a single nucleotide substitution to cause a change in the encoded amino acid. A single nucleotide substitution does not have the potential to change an amino acid to any one of the other 19. In general, its potential for change is limited to only 5 or 6 others. To evolve off the “dead point” of adaptivity, a larger step, such as the simultaneous change of more than one nucleotide, is required[8]. Moreover, the probability is close to 1 that a single mutation in a population, even though it is adaptive, will disappear without taking over the population (see my book, Chapter 3). Therefore, several adaptive mutations must occur independently and randomly at each step.
Hypermutation in the B cells does this. It quickly achieves all possible single, double, and triple mutations for the immune system, which allows them to obtain the information necessary to match a new antigen. Ordinary mutations, at the normal low rate, cannot add this information—even over long times. I shall explain why. The effects of mutation rate are nonlinear. Consider a population of antigen-activated B cells of, say, a billion individuals, which is smaller than the typical number. In two weeks, there will be about 30 generations. Let’s say the population size is stable, so in two weeks there will be a total of 30 billion replications. With a mutation rate of 1 per 1000 nucleotides per replication, there will be an average of 30 million independent changes in any particular nucleotide during a two-week period. The probability of getting two particular nucleotides to change is one per million replications. Thus in two weeks, there will be an average of 30 thousand changes in any two particular nucleotides. There will be an average of 30 changes in any three particular nucleotides. If the mutation rate is 1 per 100, these numbers would be correspondingly larger.
How many generations, and how long, would it take to get a particular multiple-nucleotide change in a germ cell to have an effect on neo-Darwinian evolution? Here, the mutation rate is about one per billion nucleotides per replication. Let’s suppose we’re doing this experiment with a population of a billion bacteria. Then, in one generation, there will be an average of one change in any particular base in some one individual. A particular double-base change has a probability of one per quintillion, or 10-18. To get one of these would take a billion generations, or about 100,000 years. To get a triple change would take 1014, or a hundred trillion, years. That is why a long waiting time cannot compensate for a low mutation rate. I’ve given numbers here for a laboratory experiment with bacteria. Many more mutations would be expected world wide. But the same kind of thing has to happen under NDT with multicelled animals as well. With vertebrates, for example, the breeding populations seldom exceed a few thousand. Multicelled animals would have many fewer mutations than those cited above for bacteria.
Max: “Your second objection to the somatic mutation model in B-cells, that ‘special enzymes’ are involved, is unsupportable. As far as I can tell from my reading of the literature, the mechanism of somatic hypermutation in B-cells is not currently known.”Spetner: On the contrary, my objection is well supported in the professional literature. The somatic hypermutations you cite do indeed require “special enzymes”, and is not the kind of mutations held to be responsible for the variation required in NDT. These mutations, unlike ordinary errors in DNA replication in the germline, are under precise control in the cell. They are turned on exactly when they are needed, and they are turned off when they have done their job. They are accurately targeted to the very small regions of the genome where they can provide variability to the CDRs, which form the antibody-binding site. They do not occur at any other place in the genome. Although the mechanism of this precisely targeted phenomenon is not yet known in complete detail, enough is known to say that there has to be a “mechanism”—hypermutation does not happen by chance. Thus, even 14 years ago, a popular textbook in cell biology said[9], “There must exist mechanisms that direct mutational activity to variable-region sequences. How this might occur is not known; possibly some sequence in the area of the variable region directs a special enzyme system to carry out point replacements of nucleotides independent of template specification.” (my emphasis)
Informed current opinion on the subject of somatic hypermutations is overwhelmingly (and perhaps even unanimously) in favor of the suggestion that they are produced by a special mechanism requiring special enzymes that are unlike the spontaneous germline mutations assumed to be responsible for evolution. Experts in this field are very clear on this point. Let me just bring you a few quotes from a recent paper by Robert Blanden and his colleagues[10], in which they describe important characteristics of somatic mutations, and note how they differ from germline mutations [all emphases are mine (LMS)]:
“The accumulated findings strongly suggest a complex mechanism [for hypermutation], which is unlikely to employ simple error-prone DNA repair processes involving DNA template directed DNA synthesis.”“… there should logically be a mechanism to ensure that when successful mutation has taken place, there is no further mutation which may destroy successful V(D)J sequences.”Let me also bring you a few quotes from another recent paper by David Winter & Patricia Gearhart (whom you may even know) on the subject of somatic hypermutations[11]:
“The pattern of somatic mutations in rearranged variable (V) genes differs from the pattern of meiotic mutations, indicating that a different mechanism generates hypermutations than generates spontaneous mutation.”“… somatic hypermutations may be derived by different mutational processes than meiotic mutations.”“The evidence suggests a model of hypermutation in which the DNA sequence of the immunoglobulin region directs the rearranged variable gene to a point on the nuclear matrix where both transcription and hypermutation occur.”“B cells that are stimulated by antigen activate an error-prone hypermutation mechanism to introduce point substitutions throughout the V region.”“… it has been shown that areas containing transcription promoters and enhancers are required for [hyper]mutation …”It thus seems quite clear that informed opinion in this field supports my contention and rejects your suggestion that “an alternative possibility is that the high rate of accumulation of mutations simply reflects selective inhibition of normal proof-reading mechanisms”. This is your field of expertise, Ed. Please let me know if you agree or disagree.
Max: “The mechanism could perhaps involve ‘special’ enzymes that create mutations, but an alternative possibility is that the high rate of accumulation of mutations simply reflects selective inhibition of normal proof-reading mechanisms. But again, I fail to see why the source of the random mutations should influence the general validity of the conclusion that random mutations and selection can increase genomic information, or why you feel that these mutations cannot serve as a model for evolutionary adaptations.”Spetner: It should be clear from what I have written above that the hypermutation in B cells cannot serve as a prototype for the random mutations required by NDT to account for evolution. There is no known mechanism for mutation in germ cells that is comparable to the hypermutation in B cells. The example of hypermutation cannot be used to support your contention that random mutations in germ cells can build up information in the genome to explain Evolution A.
Max: “… if we accept your argument against extrapolation from B cell adaptation to species adaptation, should we reject the extrapolation of any information learned from studying one organism to understand adaptations in a second organism, unless it is shown that both the rate and mechanism of mutation are the same in both organisms? In my view this would be like refusing to use the gravitational constant determined in laboratories on earth to analyze stellar physics. Such a reluctance to extrapolate would certainly prevent the use of modern organisms as a basis for understanding evolutionary events that occurred millions of years ago (which may be precisely your intent). I sometimes hear arguments like yours from creationists who are demanding rigorous ‘proof’ of evolution. These creationists do not seem to understand the distinction between mathematics, where a rigorous proof is expected, versus most experimental and observational science, where all we are seeking is the best theory that explains observed data. Of course, it is possible to extrapolate unreasonably, but I do not see that you have shown how evolutionary theory (or my essay) does this.”Spetner: Your comparison is fallacious. Extrapolations made in astrophysics and cosmology may not be entirely valid, but at least they are reasonable based on everything we know. The extrapolation you propose from B-cell hypermutation to neo-Darwinian evolution is unreasonable based on present knowledge, and it is therefore unjustified (as explained above). It is not Science.
I have not asked for a mathematical-like proof for evolution. But for heaven’s sake, how about exercising some scientific discipline? Scenarios and just-so stories cannot substitute for proof. There is no proof of Evolution A that is of a standard that would be acceptable in any other scientific area.
Max then summarized his arguments against my comments to his posting, which I reproduce below and to which I have appended my comments.
Max: “I have made these opposing points:
(1) Using your own example of streptomycin resistance, I have pointed out that some mutations of the S12 ribosomal protein do not represent a ‘loss of information’ even by your own questionable criteria.”Spetner: And I have shown above the errors of your argument. Your use of pejorative adjectives cannot make up for the weakness of your case.
Max: “(2) I have argued that your ‘information’ criteria (for deciding whether genes gain or lose information after specific mutations) are vague, non-quantitative, not supported by any logic, not accepted in the scientific literature and not demonstrably superior to other ways of judging the effects of mutation on genomic information.”Spetner: Not only have I made it clear above that my criterion for gain/loss of information is quantitative, and supported by logic and the conventional understanding of these notions in information theory, I included that section in my first critique of your posting. You chose not to relate to it at all, and instead you made up the above criticism out of thin air.
Max: “(3) I have discussed why examples of adaptive mutations in non-duplicated genes might appear to show some loss of one type of function (if not loss of information) as they gain a new function under selection by a novel environmental stress, and thus exhibit a kind of ‘bias’ that might have mislead you into making your rather risky extrapolations about the role of random mutations in evolution.
“(4) I have explained why an example of a gene duplication followed by differentiation of the two gene copies to enlarge genomic information might be hard to observe in the laboratory, contributing to the ‘bias’ mentioned above in the set of mutations that we do observe in the laboratory.”Spetner: And I have shown above that what you call a possible bias in our observations of mutations stems from your lack of understanding of my arguments. You have shown no valid reason why there is any bias in the set of all mutations that have been observed in all the genetic laboratories in the world.
Max: “(5) I have provided an example of duplicated and differentiated immunoglobulin gamma genes that can plausibly be interpreted by the evolutionary triad of mechanisms (gene duplication, random mutation and natural selection) each of which has been demonstrated individually as natural mechanism in appropriate laboratory experiments; and I have challenged you to provide an alternative more plausible explanation for the origin of these four gamma genes.”Spetner: And I have shown above that your laboratory experiments are not applicable to Evolution A. I have also pointed out that there is no obligation to provide a natural explanation of origins. There may not be one. But I encourage you to keep looking. But please remember that the solution to the problem of the origin of proteins, or the origin of life, may not be where you are looking.
Max: “(6) Finally, I have asked you to explain why hypermutation and selection of immunoglobulin genes in B cells should not serve as an instructive prototype demonstrating the potential of mutation and selection to improve function of proteins in evolution; specifically, I have asked why either the faster mutation rate in the B cell model or the unknown mechanisms of the mutations are relevant to the question of whether random mutation and natural selection can lead to increased fitness of proteins in evolution.”Spetner: And I have explained all that above and showed you why the somatic hypermutations do not qualify as examples that could pertain to the germ-line mutations required for evolution.
Summary
I have shown here, with references to my book, that the examples most often cited by evolutionists as evidence for evolution occurring now are not evidence at all for the grand sweep of evolution, which I have called here Evolution A. For an example of evolution happening now to have any relevance to Evolution A, it must be based on a mutation that could be typical of those alleged to be in the long series of steps that lead from a bacterium to a baboon. The mutation must at least be one that when repeated again and again will build up enough information to turn a bacterium into a baboon. The favorite example cited for evolution is antibiotic resistance. I have shown that the mutations leading to antibiotic resistance do not add any information to the biocosm. In some cases, they actually lose information. I have shown an example of a mutation that can easily be misconstrued to demonstrate the addition of information to the genome. Upon the gathering of further data, this example turned out to be a demonstration of information loss and not gain. Conclusion jumping is always risky, because we seldom have enough data. Yet, the evolutionist community has persisted in making the shakiest of extrapolations.Max has tried to argue that his triad of gene duplication, random mutation, and natural selection, can add information to the collective genome of the biocosm.
I have exposed his argument as being nothing more that offering possible scenarios—it is argument by just-so-stories. But the argument against NDT does not stop with the failure of its supporters to show proper theoretical or empirical evidence for it. The telling blow against NDT is that examples of information addition have never been exhibited. The absence of such examples is more than just the absence of evidence for evolution. It is actually evidence against evolution because if NDT were correct, there should be millions of such examples and in all the genetic experiments performed until now we should have seen many.
Finally, the example of mutations in the B cells of the immune system carries no weight as an example of a mutation that adds information. Although these mutations do add information to the B-cell genome, they cannot be applied to evolution for the reasons I laid out above.
Dr. Edward E. Max made a valiant attempt to present a case for evolution in his posting on the URL cited above. That he failed is not because of any defect in the author. Dr. Max is an intelligent, competent, and articulate scientist. He has a PhD and an MD, and for many years has done research and published on the genetics of the immune system, and he has made important contributions to our knowledge in this field. If he could not make a good case for evolution, there must be something woefully wrong with evolution.
Dr. Lee M. Spetner
Endnotes
[1] Wright, Sewall, (1932). “The roles of mutation, inbreeding, crossbreeding and selection in evolution,” Proceedings 6th Intnational Congress of Genetics, 1: 356-366. [RETURN TO TEXT]
[2] biocosm is a word I have coined to denote the totality of life on our planet. [RETURN TO TEXT]
[3] Burleigh, B. D., P. W. J. Rigby, & B. S. Hartley, (1974). “A comparison of wild-type and mutant ribitol dehydrogenase from Klebsiella aerogenes.” Biochem. J., 143: 341-352.. [RETURN TO TEXT]
[4] Gödel, Kurt On Formally Undecidable Propositions of Principia Mathematica and Related Systems. (1962) Translated from the German by B. Meltzer and R. B.Braithwaite. London: Oliver & Boyd. [RETURN TO TEXT]
[5] Darnell et al. (1986), Molecular Cell Biology, Scientific American Books, p. 1116. [RETURN TO TEXT]
[6] Winter, D. B. & P. J. Gearhart (1998) Dual enigma of somatic hypermutation of immunoglobulin variable genes: targeting and mechanism. Immunological Reviews 162: 89-96. [RETURN TO TEXT]
[7] For simplicity in explanation I am assuming here that the environment does not change. The same argument holds when the environment changes, with a slight alteration of the language. [RETURN TO TEXT]
[8] Evolutionists often glibly argue that a recombination in the chromosomes can provide the large change to throw the genome off the small adaptive hill it is on and provide the opportunity for it to land on another adaptive hill. But it is highly improbable that it will land at a higher adaptive elevation. This argument abandons the Darwinian premise of small, and not unlikely, changes driving evolution. [RETURN TO TEXT]
[9] Darnell, J., H. Lodish, & D. Baltimore (1986), Molecular Cell Biology, Scientific American Books, New York: Freeman. p. 1116. [RETURN TO TEXT]
[10] Blanden, R. V., H. S. Rothenfluh, P. Zylstra, G. F. Weiller, & E. J. Steele, (1998). The signature of somatic hypermutation appears to be written into the germline IgV segment repertoire. Immunological Reviews 162: 117-132. [RETURN TO TEXT]
[11] Winter, D. B. & P. J. Gearhart (1998). Dual enigma of somatic hypermutation of immunoglobulin variable genes: targeting and mechanism. Immunological Reviews 162: 89-96. [RETURN TO TEXT]
Home | Feedback | Related Links | Further Reading | Back to Top
This site has received over

visits since November 1997
© TrueOrigin Archive. All Rights Reserved. Please advise Webmaster of any technical problems.